理論¶
対称関数¶
原子が置かれている状況(具体的には、近傍の原子構造)をニューラルネットワークに入力できる形に変換するのが、対称関数です。原子のエネルギーは系の平行移動や回転、同種原子の入れ替えに対して不変(対称)であるため、エネルギーに関する特徴量を抽出する対称関数に対しても、同じ性質が要請されます。また、ニューラルネットワークの入力ノード数が固定なので、対称関数の値の数も近傍原子数に依らず一定である必要があります。例えば「近傍原子との距離」や「近傍原子のなす角度」は、平行移動や回転に対しては不変ですが、近傍原子数が増えればその数だけ値が増えるため、そのまま使うことはできません。
対称関数としては様々なものが提案されていますが、本製品ではBehlerが提案[1]した対称関数( :動径成分、
:動径成分、 または
または :角度成分):
:角度成分):
![f_c(R_{ij}) &=
\begin{cases}
\tanh^3\left[ 1 - \frac{R_{ij}}{R_c} \right] \text{or} \; \frac{1}{2}\left[ \cos\left(\frac{\pi R_{ij}}{R_c}\right)+1 \right] &\text{for} \; R_{ij}\leq R_c \\
0 &\text{for} \; R_{ij} > R_c,
\end{cases} \\
G_i^2 &= \sum_{j=1}^{N_\text{atom}} e^{-\eta (R_{ij}-R_s)^2} \cdot f_c(R_{ij}), \\
G_i^3 &= 2^{1-\zeta} \sum_{j \neq i} \sum_{k \neq i,j} \left[ (1+\lambda \cdot \cos\theta_{ijk})^\zeta \cdot e^{-\eta \left[ (R_{ij}-R_s)^2+(R_{ik}-R_s)^2+(R_{jk}-R_s)^2 \right]} \cdot f_c(R_{ij}) \cdot f_c(R_{ik}) \cdot f_c(R_{jk}) \right], \\
G_i^4 &= 2^{1-\zeta} \sum_{j \neq i} \sum_{k \neq i,j} \left[ (1+\lambda \cdot \cos\theta_{ijk})^\zeta \cdot e^{-\eta \left[ (R_{ij}-R_s)^2+(R_{ik}-R_s)^2 \right]} \cdot f_c(R_{ij}) \cdot f_c(R_{ik}) \right],](_images/math/21d32ad49346798867342323893fd48707e5acd9.svg)
Chebyshev多項式を使った対称関数[2]( :動径成分、
:動径成分、 :角度成分):
:角度成分):

および、SANNPの論文[3]で使われている対称関数( :2体成分、
:2体成分、 :3体成分):
:3体成分):

(本製品ではMany-Body対称関数と呼びます)が使用可能です。
系に異なる複数の元素が含まれる場合には、元素の組み合わせごとに対称関数 (のパラメータセット)を用意することでその違いを表現する、という方法があります:
(のパラメータセット)を用意することでその違いを表現する、という方法があります:

この方法では、系に含まれる元素の種類が増えると、その組み合わせの数だけ多数のパラメータが必要になってしまう、という問題があります。これを避けるため、対称関数は増やさずに共通のものを使い、かわりに元素ごとの「重み」を導入するという方法[2][4]が提案されています:

本製品では、Behler対称関数とChebyshev対称関数でこの重み付き対称関数が使用可能です。重みとしては、原子番号  およびその幾何平均
およびその幾何平均  を使用します。
を使用します。
ニューラルネットワーク¶
ニューラルネットワークは、入力層、1つ以上の中間層(隠れ層)、出力層からなります。各層には1つ以上のノードがあり、前の層のノードの値を入力として、次の層のノードの値を決めます。
層  のノード
のノード  の値を
の値を  とすると、
とすると、

となります。重み  、バイアス
、バイアス  がニューラルネットワークのパラメータで、求める出力を得るために、を最適化するのが「ニューラルネットワークの学習」ということになります。
がニューラルネットワークのパラメータで、求める出力を得るために、を最適化するのが「ニューラルネットワークの学習」ということになります。
重みとバイアスに加え、非線形な振る舞いを表現するために使われるのが活性化関数  です。本製品では活性化関数として
です。本製品では活性化関数として
シグモイド関数

tanh関数

twisted tanh関数

eLU関数

GELU関数

または活性化関数なし  が選べます。
が選べます。
SANNP¶
構造中のある原子 に対して、近傍の構造から対称関数を計算し、ニューラルネットワークに入力すると、出力としてその原子のエネルギー  が得られます。
が得られます。
一方、教師データとしては密度汎関数理論(DFT)に基づく第一原理計算が使われますが、その結果は「各原子のエネルギー」という形にはなっていません。系の全エネルギー  を使い、
を使い、  を残差として最適化を行う方法がありますが、この場合1つの原子構造に対するDFT計算からエネルギーに関する情報は1つしか得られないことになります。
を残差として最適化を行う方法がありますが、この場合1つの原子構造に対するDFT計算からエネルギーに関する情報は1つしか得られないことになります。
本製品で採用しているSANNP(Single Atom Neural Network Potential)[3]では、DFT計算の結果を各原子のエネルギー  に分割する手法[5]を使うことで、各原子のエネルギーを残差
に分割する手法[5]を使うことで、各原子のエネルギーを残差  を使って直接最適化しています。これにより、1つの原子構造に対するDFT計算から得られるエネルギーに関する情報が原子数倍になり、少ないDFT計算の結果からでも効率よく学習を行うことができます。
を使って直接最適化しています。これにより、1つの原子構造に対するDFT計算から得られるエネルギーに関する情報が原子数倍になり、少ないDFT計算の結果からでも効率よく学習を行うことができます。
また、エネルギーと同様に、ニューラルネットワークを使って「各原子の電荷」を得ることができます。クーロン相互作用は長距離でも働くため、クーロン相互作用も含めてニューラルネットワーク力場で計算しようとすると大きなカットオフ半径が必要になります。本製品ではニューラルネットワーク力場と、ニューラルネットワークで計算した電荷を使ったクーロン相互作用を組み合わせて使うことができますので、短距離の相互作用を扱うニューラルネットワーク力場の部分についてはカットオフ半径を小さくして計算することが可能です。
系の全エネルギーは、ニューラルネットワークで計算した各原子のエネルギー を使って、電荷を使わない場合

と表現されます。また、ニューラルネットワークで計算した電荷  を使う場合は、系の全電荷が0になるようシフトした上で、
を使う場合は、系の全電荷が0になるようシフトした上で、
![E_\text{tot}^\text{NN} &= E_\text{short} + E_\text{elec} \\
&= \sum_i E_i^\text{NN} + \sum_i \sum_{j>i}^{R_{ij} < R_\text{elec}} \frac{Q_i^\text{NN} Q_j^\text{NN}}{4\pi\epsilon_0R_{ij}} \cdot f_\text{screen}(R_{ij}), \\
f_\text{screen}&(R_{ij}) =
\begin{cases}
\frac{1}{2}\left[1-\cos\left(\frac{\pi \cdot R_{ij}}{R_\text{short}}\right)\right] \; & \text{for} \; R_{ij} \leq R_\text{short} \\
1 & \text{for} \; R_{ij} > R_\text{short}
\end{cases}](_images/math/4f0c3943b3cb03600efad60a06870f562489fa70.svg)
として計算します。
HDNNPにおける原子エネルギー推定法¶
NNPを最適化する際に、原子エネルギーの平均値および分散はニューラルネットワークの最終層の初期推定を行うのに重要な情報となります。第一原理計算によって得られる情報は、使用する計算手法(DFT+UやHybrid汎関数など)によっては十分な精度が得られず、ニューラルネットワークの学習の収束性が悪化してしまいます。また、VASP等の別のソフトウェアの結果を変換して教師データを得た場合にも、原子エネルギーのデータがないため、収束性が低下してしまいます。
そこで、HDNNP(教師データとして系の全エネルギーを使う従来のNNP)使用時には、全エネルギーと化学量論係数から成る連立方程式を解いて原子エネルギーの平均値と分散を計算します。具体的には、下式を解きます。

 は各元素種の原子エネルギーの平均値(ベクトル)、
は各元素種の原子エネルギーの平均値(ベクトル)、 は各教師データにおける全エネルギー(ベクトル)、
は各教師データにおける全エネルギー(ベクトル)、 は各教師データにおける化学量論係数(教師データ数×元素種数の行列)です。
は各教師データにおける化学量論係数(教師データ数×元素種数の行列)です。 の一般化逆行列を左から両辺に作用させるとが求まります。分散についても同様の式を解きます。線形従属で無い限り、SANNPの原子エネルギーと概ね同じくらいのオーダーの値が得られる傾向にあります。
の一般化逆行列を左から両辺に作用させるとが求まります。分散についても同様の式を解きます。線形従属で無い限り、SANNPの原子エネルギーと概ね同じくらいのオーダーの値が得られる傾向にあります。
メトロポリス法による構造生成¶
元となる構造からランダムに原子を変位させて教師データ用の構造を生成する場合、その結果として生じる対称関数の分布、あるいはエネルギーの分布については考慮されません。そのため、構造自体は満遍ないように見えても、ある対象関数の範囲での学習が不十分になってしまう、ということがあります。
本製品では、メトロポリス法による構造生成を行う機能があります。メトロポリス法では、確率的な遷移過程を使って、ボルツマン分布に従うエネルギー分布を持つように構造を生成することができます。
最初に元となる構造を考え、その構造を変化(原子の変位、または入れ替え)させて遷移先の候補となる新しい構造を作ります。学習済みのNNPを使って、それぞれの構造のエネルギー 、
、 を計算します。エネルギーの差
を計算します。エネルギーの差  から、遷移確率を次のように決めます:
から、遷移確率を次のように決めます:

この確率によって、新しい構造に遷移する(採択)か、遷移しない(棄却)かを決めます。遷移したら、遷移先の構造を元として、また次の候補となる構造を作ります。
この手順を繰り返していくと、温度 でのボルツマン分布に従うようなエネルギー分布を持つ構造を生成することができます。この構造を元に教師データを作り、ニューラルネットワークを再度学習させる(強化学習)ことで、より広いエネルギーの構造をカバーするNNPを作ることができます。
でのボルツマン分布に従うようなエネルギー分布を持つ構造を生成することができます。この構造を元に教師データを作り、ニューラルネットワークを再度学習させる(強化学習)ことで、より広いエネルギーの構造をカバーするNNPを作ることができます。
Δ-NNP¶
全エネルギーをNNPで表現するのではなく、第0近似として古典力場による2体間エネルギーを使い、そこからの差分をNNPを使って表現する、という方法です。古典力場としては、通常のLJ力場に加えて次数の異なる項を増やしたLJ-like力場を使っています。

古典力場のパラメータについては、教師データを使ってあらかじめ最適化しておきます。ニューラルネットワークを学習する際の教師データには、DFT計算と古典力場のエネルギーの差分  を使います。
を使います。
Δ-NNPは通常のNNPに比べてロバストで、少ない教師データで作成した力場でも、大きな破綻が起きにくいのが特長です。外挿も機能し、例えば300 Kの教師データで作成した力場でも、1000 Kでそれなりに上手く動きます。
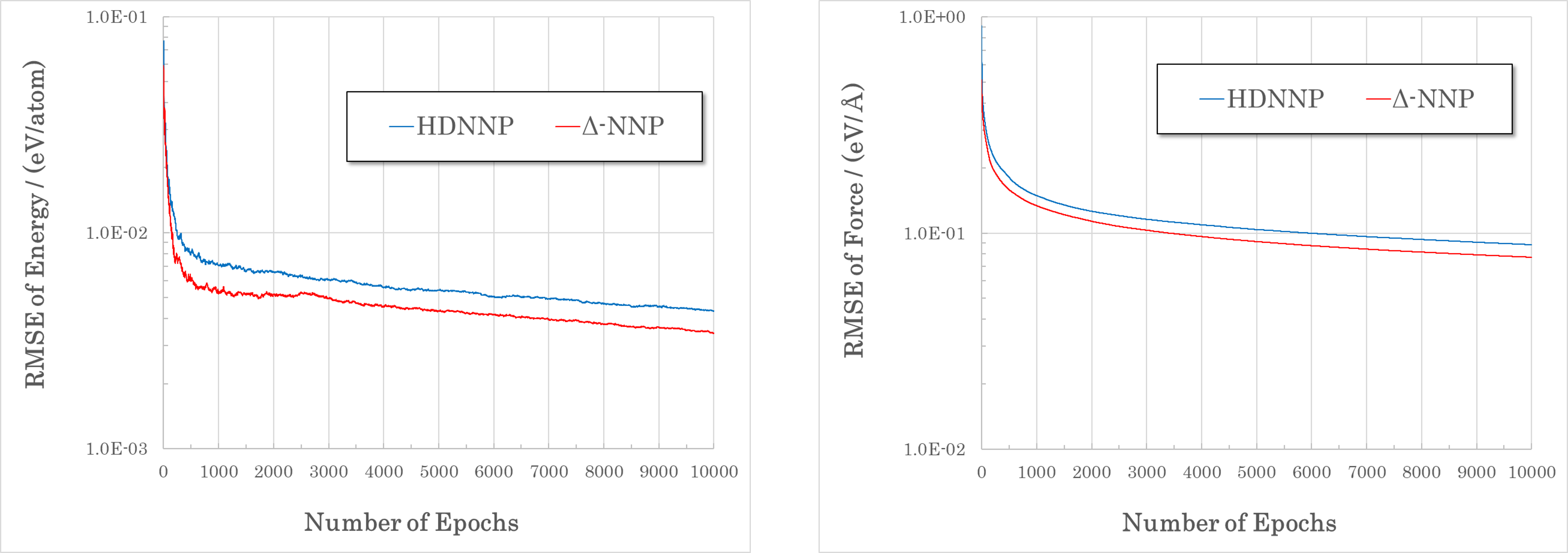
図: Δ-NNPとHDNNPを同じ教師データ(Li10GeP2S12、6914構造)を使って学習し、RMSEを比較しました。Δ-NNPはHDNNPよりも収束性が良いことが確認できます。¶
ReaxFFを用いたΔ-NNP¶
Δ-NNPの古典力場としてReaxFF[7]を使う方法です。ReaxFFは多体力場であり、LJ(-like)力場に比べてよりよくエネルギー・力を表現することができます。本製品ではReaxFFのエネルギーとして結合エネルギー( )、Over-coordinationエネルギー(
)、Over-coordinationエネルギー( )、Under-coordinationエネルギー(
)、Under-coordinationエネルギー( )、Lone-pairエネルギー(
)、Lone-pairエネルギー( )、van der Waalsエネルギー(
)、van der Waalsエネルギー( )のみを使用します。ReaxFFのパラメータは既存のものを使用し、本製品での最適化は行いません。ReaxFFの寄与を係数(混合率)
)のみを使用します。ReaxFFのパラメータは既存のものを使用し、本製品での最適化は行いません。ReaxFFの寄与を係数(混合率)  で指定し、そこからの差分をNNPを使って表現します。
で指定し、そこからの差分をNNPを使って表現します。

自己学習ハイブリッドモンテカルロ法¶
自己学習ハイブリッドモンテカルロ(Self-learning hybrid Monte Carlo, SLHMC)法[6]は、教師データの生成とニューラルネットワーク力場の学習を同時に進めることができる手法です。以下のようなアルゴリズムに従って実行します。
初期構造から第一原理分子動力学計算をある程度進め、それを教師データとして初期ニューラルネットワーク力場を作る。
ニューラルネットワーク力場による分子動力学計算(NVE)で構造を時間発展させる(
 )。
)。発展後の構造
 について第一原理計算でエネルギー
について第一原理計算でエネルギー  を計算する。
を計算する。以下の遷移確率
 を使って、メトロポリス法により構造を採択するかどうか決める。
を使って、メトロポリス法により構造を採択するかどうか決める。
棄却する場合は、構造を
 に戻し、運動量
に戻し、運動量  を温度 におけるボルツマン分布で定義しなおす。
を温度 におけるボルツマン分布で定義しなおす。1.~3.をある程度繰り返したら、生成された構造(採択された構造のみ、または棄却されたものも含めた全ての構造)を教師データとしてニューラルネットワーク力場の再学習を行う。
1.~3.と4.を繰り返す。
この方法では、教師データの準備すら行う必要がなく、1つの初期構造を用意するだけで後は自動的にニューラルネットワーク力場を作ることができます。力場作成の手間が大幅に削減でき、また教師データの質など手順に依存する要素が排除されるため、常に同水準の力場を作ることができます。教師データの数も低減できる傾向にあるため、計算に要する時間も大幅に削減できます。
モンテカルロ法の遷移確率(3.)は第一原理計算により決めているため、教師データのアンサンブルが厳密に第一原理分子動力学計算のアンサンブルと一致するということもこの方法の特長です。
また、本製品では構造の時間発展(1.)の前にNPHによるセルの変形を行う、または構造の時間発展(1.)の分子動力学計算をNVEではなくNPHで行うこともできます。これによりセル形状の異なる教師データも含まれるようになり、格子定数(密度)が未知の物質に対しても本手法を使って力場を作ることができるようになります。この場合、厳密なモンテカルロ法ではなくなりますが、力場を作るという目的の上では問題なく使えます。